
医薬品を欧州市場に持ち込むには、規制当局への提出を準備する以上のことが必要です。企業は、製品の承認、品質基準、ライフサイクル管理、および欧州全体のコンプライアンス義務に影響を与える機関や組織のネットワークを理解する必要があります。
欧州医薬品庁(EMA)は多くの医薬品承認の中心であり続ける一方で、欧州の規制枠組みは単一の権限を超えて広がっています。医薬品庁長官(HMA)、欧州医薬品・ヘルスケア品質局(EDQM)、スイスメディック、医薬品・ヘルスケア製品規制庁(MHRA)などの組織は、医薬品がライフサイクルを通じてどのように審査、監視、維持されるかを形作る上でそれぞれ重要な役割を果たしています。これらの組織は一体となって、欧州の規制が独立国家システムから、規制の監視、品質基準、市場アクセスのバランスをとる協調的なエコシステムへとどのように進化してきたかを示しています。
これらの組織は一体となって、欧州の医薬品規制が独立国家システムの集合から、国固有の監督と調和することのバランスをとる、高度に調整された規制エコシステムへとどのように進化したかを説明しています。
欧州の規制枠組みの進化
1990年代以前は、ヨーロッパでの医薬品承認は主に個々の国家当局によって管理されていた。各国は独自の規制手順、審査基準、および承認タイムラインを維持し、複数の欧州市場へのアクセスを求める企業に多大な複雑さをもたらしました。
医薬品開発がますますグローバル化するにつれて、規制当局は、より一層の協力、より強力な品質基準、より効率的な審査プロセスの必要性を認識しました。これにより、地域全体の調整、調和、規制の一貫性をサポートするよう設計された複数の組織の作成と拡大につながりました。
EMAの設立により、特定の医薬品の中央審査経路が導入された。同時に、HMAのような協力体制は国家当局間の協力を強化し、EDQMは欧州薬局方及び適合証明書(CEP)システムを通じて医薬品品質基準の調和の役割を拡大した。
最近では、EUの枠組み外でMHRAの独立した役割を創出することで、ブレグジットなどの規制の進展が状況をさらに変えました。
今日、製薬会社は、複数の機関や組織を含む規制環境をナビゲートしなければならず、それぞれに明確な責任と規制上の影響があります。 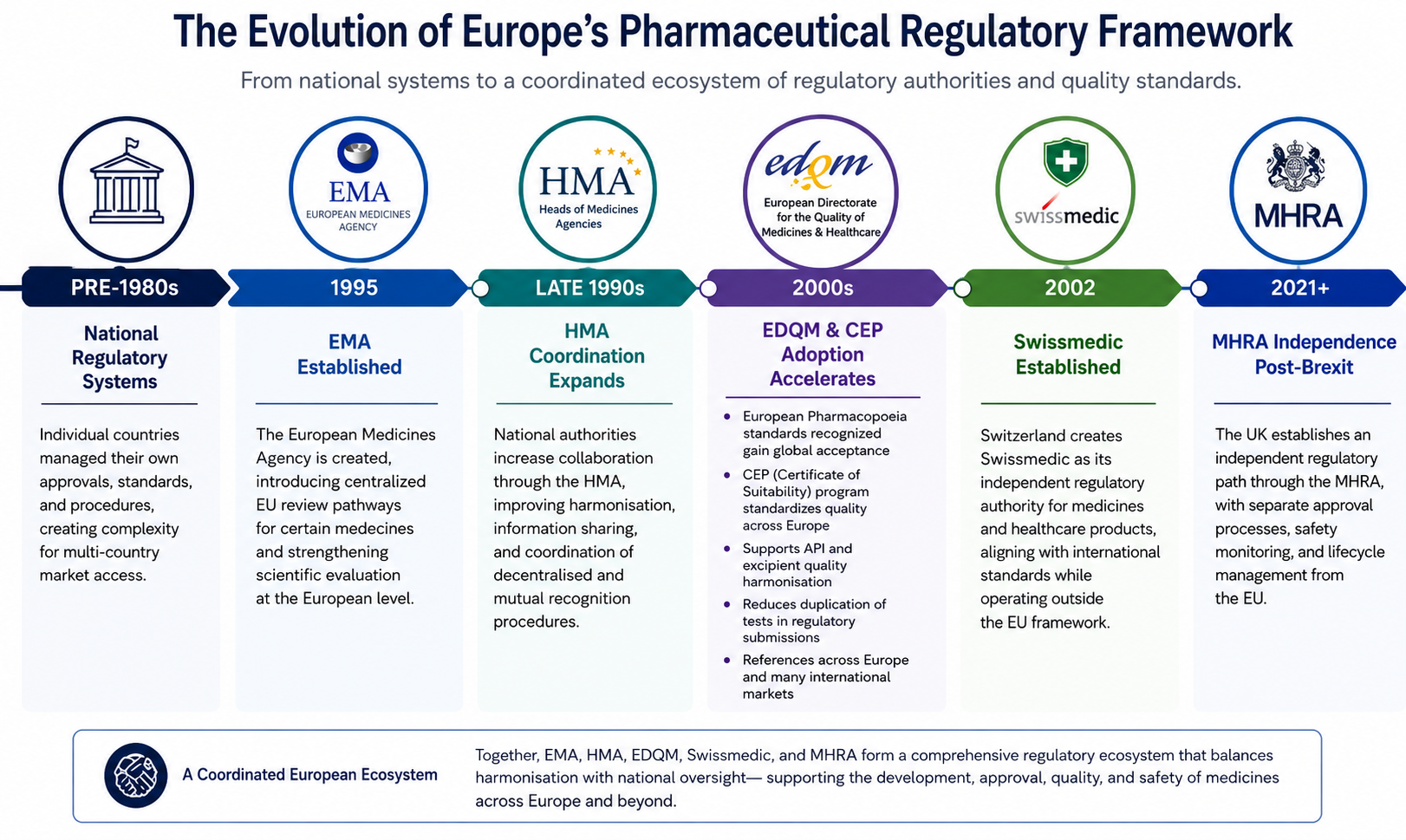
医薬品庁長官の役割(HMA)
医薬品庁長官(HMA)は、欧州経済領域(EEA)全域で医薬品規制を担当する国の所轄官庁で構成されるネットワークです。
EMAとは異なり、HMAは一元的な承認権限ではありません。代わりに、ヨーロッパの国家機関間の規制活動を調整するのに役立つ協力的な枠組みとして機能します。
HMAは1990年代に設立され、欧州の規制協力が分散型および相互認証手続きの導入とともに拡大した。複数の国を同時に巻き込む医薬品申請が増えるにつれて、規制当局は国家当局間のより強力な調整の必要性を認識しました。
時間の経過とともに、HMAはヨーロッパの規制構造の重要な部分となり、以下を改善しています。
- 規制の調和
- 当局間の情報共有
- 分散型処置中の調整
- ファーマコビジランスのコラボレーション
- 規制データの標準化
また、欧州の規制活動全体の一貫性の向上に重点を置いたワーキンググループやイニシアチブもサポートしています。
製薬会社にとって、HMAは、欧州全体の科学的評価、手順基準、規制上の期待に国家機関が一致させるのを支援する上で、舞台裏で重要な役割を果たしています。
この調整は、特に以下の場合に重要になります。
- 分散型手順(DCP)
- 相互認識手順(MRP)
- 多国間ライフサイクル管理活動
- 国境を越えた医薬品安全性監視の調整
HMAの枠組みを理解することで、企業は欧州の審査プロセスを通じて各国当局がどのように協力するかをより良く予測することができます。
EDQMおよびCEP:医薬品品質基準の基礎
EMA、HMA、Swissmedic、MHRAなどの機関は規制監督と市販承認活動に重点を置いていますが、欧州医薬品・ヘルスケア品質局(EDQM)は、異なるが同様に重要な機能を果たしています。
欧州評議会の下で運営されているEDQMは、世界で最も広く認知されている医薬品品質基準のコレクションの一つである欧州薬局方の開発と維持に責任を持つ独立した欧州の機関です。
医薬品規制に対するEDQMの最も影響力のある貢献の1つは、適合証明書(CEP)プログラムです。
CEPは、関連する欧州薬局方モノグラフの要件に従って、医薬品の品質を適切に管理できることを実証する。製造業者は、広範な物質データを複数の規制当局に繰り返し提出するのではなく、規制当局への提出書類の中でCEPを参照することができます。
時間の経過とともに、CEPは欧州の医薬品規制の基礎となり、以下によって頻繁に利用されています。
- 原薬(API)製造業者• 添加剤製造業者(該当する場合)・製造販売業者• 欧州全域の規制当局• 国際的な規制当局への提出を求める企業
CEPの使用の増加は、医薬品品質基準の調和強化に向けた欧州規制の重要な変化を反映している。複雑なサプライチェーンや複数市場への提出を管理する企業にとって、CEPは一貫性の向上、重複の削減、より効率的な規制審査プロセスのサポートに役立ちます。
Swissmedic – スイスの独立規制アプローチ
スイスは欧州連合の加盟国ではありませんが、Swissmedicはヨーロッパで最も尊敬されている医薬品規制当局の1つです。
Swissmedicは、スイスの地域の規制監督システムを中央の国家機関に統合した後、2002年に設立されました。それ以来、EUシステム外の独立した規制枠組みを維持しながら、国際協力の取り組みを拡大し続けています。
今日、Swissmedicは、以下の責任を担っています。
- スイスでの医薬品の審査と承認
- 製品の安全性の監視
- 検査の実施
- 製造コンプライアンスの監督
- 医薬品安全性監視活動の管理
スイスは、引き続き世界的な産業プレゼンスを持つ重要な医薬品市場であり、Swissmedicは、より広範な欧州の商業化戦略を追求する企業にとって重要な権限となっています。
Swissmedicは国際的な規制当局と協力し、信頼または認識の道筋に参加する可能性がありますが、審査手順はEMAから独立しています。
製薬会社にとって、これはスイスの申請には、多くの場合、以下の点について慎重な計画が必要であることを意味します。
- 国別の行政要件
- 現地での代理義務
- 製品のラベル表示に関する期待事項
- スイス固有の提出スケジュール
- 言語および文書の要件
企業は、EUの承認が自動的にスイスの規制上の期待との整合性を保証すると仮定することは避けるべきです。EU市場とスイス市場の両方で組織的な提出戦略を維持することは、効率的な市場アクセスに不可欠です。
MHRA – 英国の規制移行
医薬品・医療製品規制庁(MHRA)は、英国の医薬品、医療機器、医療製品に関する国の規制当局です。
MHRAは長い間、欧州の医薬品規制において重要な役割を果たしてきました。しかし、その責任は英国が欧州連合から離脱した後に大きく進化した。
ブレグジット以前は、多くの英国の医薬品承認はより広範なEMAフレームワーク内で行われていました。MHRAは独立した規制当局になって以来、英国市場における規制の柔軟性をサポートするように設計された、個別の承認経路、独立したライフサイクル管理手順、および新しい国際的な依存モデルを導入してきました。
現在、MHRAは以下を監督しています。
- 英国での製造販売承認
- 医薬品の安全性とファーマコビジランス
- GMP検査
- 治験の監視
- 規制コンプライアンスの監視
製薬企業にとって、BrexitはEUと英国市場の間で全く新しい規制ダイナミクスを生み出しました。
EUと英国の商業化戦略を管理する組織は、以下を維持する必要があります。
- 個別の規制当局への提出書類
- 独立したライフサイクル管理活動
- ラベル表示のコンプライアンス
- 英国固有の規制スケジュール
- パラレル医薬品安全性監視の調整
同時に、MHRAは、規制上の監視を維持しながら医薬品へのアクセスを加速することを目的とした国際的な協力と依存の経路の開発を続けています。
この進化する環境により、両法域で事業を展開する企業にとって、規制の強力な調整がますます重要になっています。
これらの権限の理解が重要な理由
HMA、Swissmedic、MHRAの運用は異なりますが、それぞれ欧州の広範な医薬品規制環境において重要な役割を果たしています。
これらの当局がどのように進化し、今日どのように機能するかを理解することは、製薬会社に以下のことに役立ちます。
- より強力な市場参入戦略を構築する
- より整理された提出物を準備する
- 国別の期待を予測する
- ライフサイクル管理の調整を改善する
- 回避可能な提出の遅延を軽減
製薬規制がグローバル市場全体でますます相互接続されるにつれて、企業は地域的な調和と国別のコンプライアンス要件のバランスを取る必要があります。
構造化された規制戦略がなければ、代理店間の違いは、管理上の非効率性、一貫性のない文書、運用の複雑さの増加につながる可能性があります。
マルチエージェンシー規制管理における一般的な課題
複数の欧州当局への提出を管理するには、多くの場合、企業が以下を調整する必要があります。
- 異なる規制タイムライン
- 国別の管理文書
- 製品情報の更新
- ファーマコビジランスの義務
- ライフサイクル管理活動
- 提出のフォーマットに関する期待事項
企業が欧州および国際市場に進出するにつれて、規制記録の一貫性を維持することがますます重要になってきています。提出、スケジュール、または管理上の要件の間にわずかな違いがあっても、回避可能な遅延や規制上の圧力が生じる可能性があります。
適切に整理された提出プロセスは、規制当局間の不整合のリスクを低減しながら、規制活動全体に対する可視性を高めるのに役立ちます。
多くの製薬企業にとって、これらの責任を社内で管理することは、特に複数の市場、進化する規制上の期待、および継続的なライフサイクル活動が申請に含まれる場合、リソース集約的になる可能性があります。
欧州の進化する規制状況のナビゲート
経験豊富なグローバル規制エキスパートであるRegistar Corpは、製薬会社が欧州の規制サポートサービスを通じて、より明確かつ自信を持って複雑な欧州の規制活動に取り組むお手伝いをします。
当社のチームは、以下を通じて企業をサポートします。
- 複数国への提出管理
- eCTD出版サポート
- 管理上の更新とライフサイクル活動
- 規制文書組織
- ASMFおよびCEP管理サポート
当社の規制専門家は、文書の一貫性を向上させ、より効率的な規制調整をサポートし、企業が規制文書と手続きワークフローをより強力に管理できるようにすることで、申請準備の簡素化を支援します。
また、重要な規制関連文書や提出記録への集中アクセスの維持を支援するために設計されたオンライン文書リポジトリであるRegistarHubへの安全なアクセスも提供しています。
欧州および国際的な規制の枠組みが進化し続ける中、構造化された規制プロセスに投資し、経験豊富な申請サポートを提供する企業は、コンプライアンスを維持し、管理の複雑さを軽減し、複数の法域にわたる持続可能な市場成長をサポートする立場にあります。
