
Portare un prodotto farmaceutico sul mercato europeo richiede molto di più che preparare una presentazione normativa. Le aziende devono comprendere la rete di agenzie e organizzazioni che influenzano le approvazioni dei prodotti, gli standard di qualità, la gestione del ciclo di vita e gli obblighi di conformità in tutta Europa.
Mentre l’Agenzia europea per i medicinali (EMA) rimane centrale per molte approvazioni farmaceutiche, il quadro normativo europeo si estende ben oltre un’unica autorità. Organizzazioni come i Heads of Medicines Agencies (HMA), la Direzione europea per la qualità dei medicinali e dell’assistenza sanitaria (EDQM), Swissmedic e l’Agenzia di regolamentazione dei medicinali e dei prodotti sanitari (MHRA) svolgono un ruolo importante nel definire il modo in cui i farmaci vengono esaminati, monitorati e mantenuti per tutto il loro ciclo di vita. Insieme, queste organizzazioni illustrano come la regolamentazione europea si è evoluta da sistemi nazionali indipendenti in un ecosistema coordinato che bilancia la supervisione normativa, gli standard di qualità e l’accesso al mercato.
Insieme, queste organizzazioni illustrano come la regolamentazione farmaceutica europea si è evoluta da una raccolta di sistemi nazionali indipendenti in un ecosistema normativo altamente coordinato che bilancia l’armonizzazione con la supervisione specifica del paese.
L’evoluzione del quadro normativo europeo
Prima degli anni ’90, le approvazioni farmaceutiche in Europa erano gestite principalmente da singole autorità nazionali. Ogni paese ha mantenuto le proprie procedure normative, gli standard di revisione e le tempistiche di approvazione, creando una complessità significativa per le aziende che cercano l’accesso a più mercati europei.
Man mano che lo sviluppo farmaceutico diventava sempre più globale, le autorità di regolamentazione riconoscevano la necessità di una maggiore cooperazione, standard di qualità più forti e processi di revisione più efficienti. Ciò ha portato alla creazione e all’espansione di diverse organizzazioni progettate per supportare il coordinamento, l’armonizzazione e la coerenza normativa in tutta la regione.
L’istituzione dell’EMA ha introdotto percorsi di revisione centralizzati per alcuni farmaci. Allo stesso tempo, strutture collaborative come l’HMA hanno rafforzato la cooperazione tra le autorità nazionali, mentre l’EDQM ha ampliato il ruolo degli standard di qualità farmaceutica armonizzati attraverso il sistema europeo di farmacopea e certificato di idoneità (CEP).
Più di recente, gli sviluppi normativi come la Brexit hanno ulteriormente rimodellato il panorama creando un ruolo indipendente per l’MHRA al di fuori del quadro dell’Unione europea.
Oggi, le aziende farmaceutiche devono navigare in un ambiente normativo che include più agenzie e organizzazioni, ognuna con responsabilità distinte e influenza normativa. 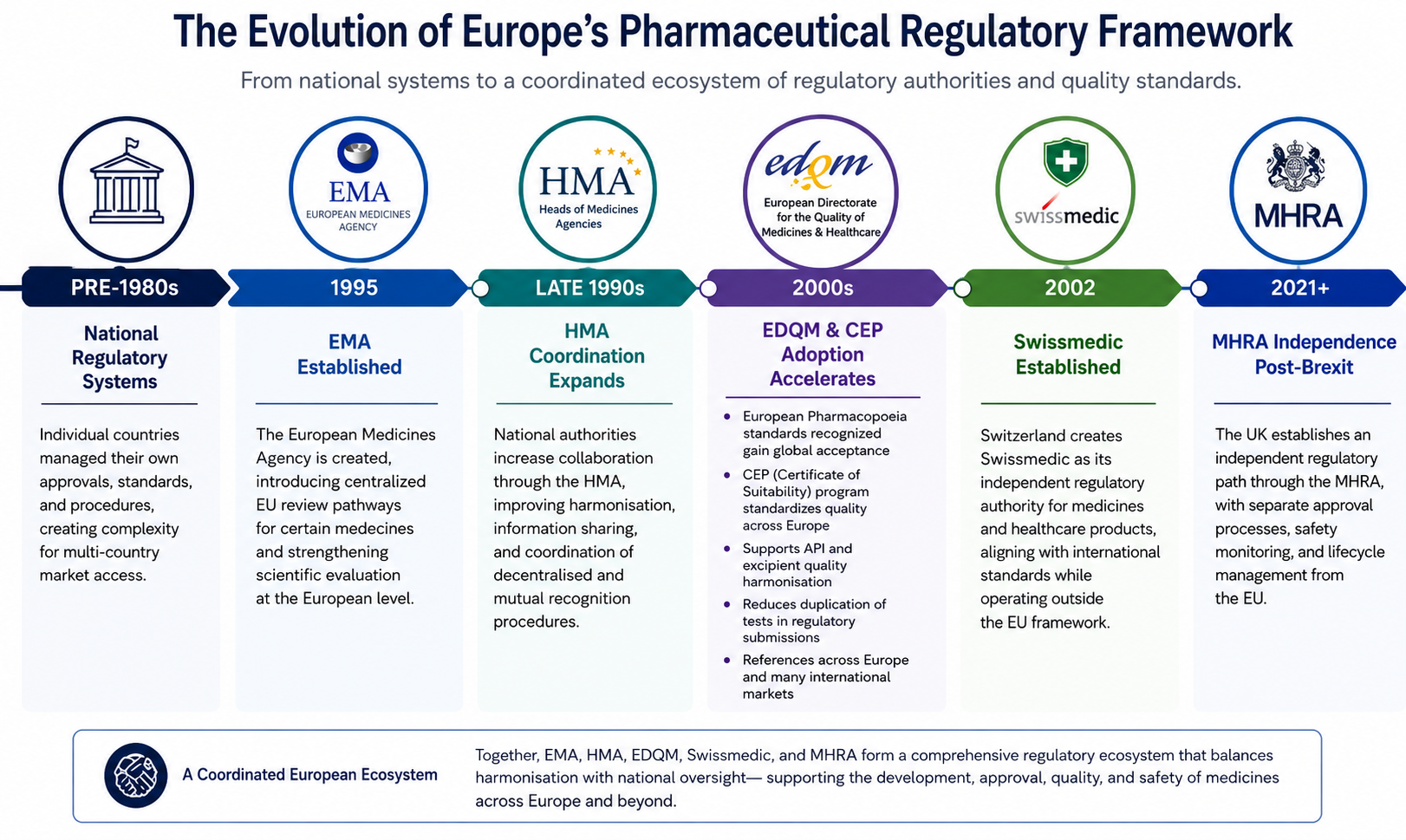
Il ruolo dei responsabili delle agenzie per i medicinali (HMA)
I capi delle agenzie per i medicinali (HMA) sono una rete composta dalle autorità nazionali competenti responsabili della regolamentazione dei medicinali in tutto lo Spazio economico europeo (SEE).
A differenza dell’EMA, l’HMA non è un’autorità di approvazione centralizzata. Invece, funge da quadro collaborativo che aiuta a coordinare le attività di regolamentazione tra le agenzie nazionali europee.
L’HMA è stata formalmente istituita negli anni ’90, con l’espansione della cooperazione normativa europea e l’introduzione di procedure di riconoscimento decentralizzate e reciproche. Man mano che iniziavano a interessare più paesi contemporaneamente, le autorità di regolamentazione riconoscevano la necessità di un maggiore coordinamento tra le autorità nazionali.
Nel corso del tempo, l’HMA è diventata una parte importante della struttura normativa europea contribuendo a migliorare:
- Armonizzazione normativa
- Condivisione delle informazioni tra le autorità
- Coordinamento durante procedure decentralizzate
- Collaborazione per la farmacovigilanza
- Standardizzazione dei dati normativi
L’organizzazione sostiene anche gruppi di lavoro e iniziative incentrate sul miglioramento della coerenza tra le attività normative europee.
Per le aziende farmaceutiche, l’HMA svolge un ruolo importante dietro le quinte nell’aiutare le agenzie nazionali ad allinearsi su valutazioni scientifiche, standard procedurali e aspettative normative in tutta Europa.
Questa coordinazione diventa particolarmente importante durante:
- Procedure decentralizzate (DCP)
- Procedure di riconoscimento reciproco (MRP)
- Attività di gestione del ciclo di vita in più Paesi
- Coordinamento transfrontaliero per la farmacovigilanza
Comprendere il quadro HMA può aiutare le aziende ad anticipare meglio il modo in cui le autorità nazionali collaborano durante i processi di revisione europei.
EDQM e CEP: La base degli standard di qualità farmaceutica
Mentre agenzie come EMA, HMA, Swissmedic e MHRA si concentrano sulle attività di supervisione normativa e autorizzazione al mercato, la Direzione europea per la qualità dei medicinali e dell’assistenza sanitaria (EDQM) svolge una funzione diversa ma altrettanto importante.
Operando nell’ambito del Consiglio d’Europa, EDQM è un’istituzione europea indipendente responsabile dello sviluppo e del mantenimento della Farmacopea europea, una delle collezioni di standard di qualità farmaceutica più ampiamente riconosciute al mondo.
Uno dei contributi più influenti di EDQM alla regolamentazione farmaceutica è il programma Certificato di Idoneità (CEP).
Un CEP dimostra che la qualità di una sostanza farmaceutica può essere adeguatamente controllata in base ai requisiti della relativa monografia della Farmacopea europea. Piuttosto che inviare ripetutamente dati estesi sulle sostanze a più autorità normative, i produttori possono fare riferimento a un CEP all’interno delle loro presentazioni normative.
Nel corso del tempo, i CEP sono diventati una pietra miliare della regolamentazione farmaceutica europea e sono spesso utilizzati da:
- Produttori di principi attivi farmaceutici (API)• Produttori di eccipienti (ove applicabile)• Titolari dell’autorizzazione all’immissione in commercio• Autorità di regolamentazione in tutta Europa• Aziende che perseguono richieste normative internazionali
Il crescente utilizzo dei CEP riflette un importante cambiamento nella regolamentazione europea verso una maggiore armonizzazione degli standard di qualità farmaceutica. Per le aziende che gestiscono catene di fornitura complesse e presentazioni multi-mercato, i CEP possono contribuire a migliorare la coerenza, ridurre la duplicazione e supportare processi di revisione normativa più efficienti.
Swissmedic – Approccio normativo indipendente della Svizzera
Sebbene la Svizzera non sia membro dell’Unione europea, Swissmedic rimane una delle autorità di regolamentazione farmaceutica più rispettate d’Europa.
Swissmedic è stata fondata nel 2002 a seguito del consolidamento dei sistemi di vigilanza regionale della Svizzera in un’autorità nazionale centralizzata. Da allora, l’agenzia ha continuato ad espandere i suoi sforzi di cooperazione internazionale mantenendo un quadro normativo indipendente al di fuori del sistema dell’UE.
Oggi Swissmedic è responsabile di:
- Revisione e autorizzazione di medicinali in Svizzera
- Monitoraggio della sicurezza del prodotto
- Esecuzione delle ispezioni
- Supervisionare la conformità della produzione
- Gestione delle attività di farmacovigilanza
La Svizzera rimane un importante mercato farmaceutico con una forte presenza a livello mondiale, rendendo Swissmedic un’autorità chiave per le aziende che perseguono strategie di commercializzazione europee più ampie.
Sebbene Swissmedic collabori con le autorità di regolamentazione internazionali e possa partecipare a percorsi di affidamento o riconoscimento, le sue procedure di revisione rimangono indipendenti dall’EMA.
Per le aziende farmaceutiche, ciò significa che le presentazioni svizzere richiedono spesso un’attenta pianificazione:
- Requisiti amministrativi specifici per Paese
- Obblighi di rappresentanza locale
- Aspettative di etichettatura del prodotto
- Tempistiche di presentazione specifiche per la Svizzera
- Requisiti di lingua e documentazione
Le aziende dovrebbero evitare di presumere che l’approvazione dell’UE garantisca automaticamente l’allineamento alle aspettative normative svizzere. Il mantenimento di strategie di presentazione organizzate in entrambi i mercati dell’UE e della Svizzera rimane essenziale per un accesso efficiente al mercato.
MHRA – Transizione normativa del Regno Unito
L’Agenzia di regolamentazione dei medicinali e dei prodotti sanitari (MHRA) funge da autorità nazionale di regolamentazione del Regno Unito per medicinali, dispositivi medici e prodotti sanitari.
L’MHRA ha svolto a lungo un ruolo importante nella regolamentazione farmaceutica europea. Tuttavia, le sue responsabilità si sono evolute in modo significativo dopo la partenza del Regno Unito dall’Unione europea.
Prima della Brexit, molte approvazioni farmaceutiche del Regno Unito operavano all’interno del più ampio quadro EMA. Da quando è diventata un’autorità di regolamentazione indipendente, l’MHRA ha introdotto percorsi di approvazione separati, procedure di gestione indipendente del ciclo di vita e nuovi modelli di affidamento internazionali progettati per supportare la flessibilità normativa all’interno del mercato britannico.
Oggi, l’MHRA sovrintende a:
- Autorizzazioni all’immissione in commercio nel Regno Unito
- Sicurezza e farmacovigilanza dei farmaci
- Ispezioni GMP
- Supervisione della sperimentazione clinica
- Monitoraggio della conformità normativa
Per le aziende farmaceutiche, la Brexit ha creato una dinamica normativa completamente nuova tra i mercati dell’UE e del Regno Unito.
Le organizzazioni che ora gestiscono le strategie di commercializzazione sia dell’UE che del Regno Unito devono mantenere:
- Presentazioni normative separate
- Attività di gestione indipendente del ciclo di vita
- Conformità all’etichettatura distinta
- Tempistiche normative specifiche per il Regno Unito
- Coordinamento parallelo della farmacovigilanza
Allo stesso tempo, l’MHRA continua a sviluppare percorsi di collaborazione e affidamento internazionali volti ad accelerare l’accesso ai farmaci, pur mantenendo la supervisione normativa.
Questo ambiente in continua evoluzione ha reso sempre più importante un forte coordinamento normativo per le aziende che operano in entrambe le giurisdizioni.
Perché la comprensione di queste autorità è importante
Sebbene HMA, Swissmedic e MHRA operino in modo diverso, ciascuno svolge un ruolo importante nel più ampio contesto normativo farmaceutico europeo.
Capire come queste autorità si sono evolute e come funzionano oggi aiuta le aziende farmaceutiche:
- Costruire strategie di ingresso sul mercato più forti
- Preparare presentazioni più organizzate
- Anticipare le aspettative specifiche del Paese
- Migliorare il coordinamento della gestione del ciclo di vita
- Ridurre i ritardi evitabili nell’invio
Man mano che la regolamentazione farmaceutica diventa sempre più interconnessa tra i mercati globali, le aziende devono bilanciare l’armonizzazione regionale con i requisiti di conformità specifici per paese.
Senza una strategia normativa strutturata, le differenze tra le agenzie possono creare inefficienze amministrative, documentazione incoerente e maggiore complessità operativa.
Sfide comuni nella gestione normativa multi-agenzia
La gestione delle presentazioni tra più autorità europee richiede spesso alle aziende di coordinare:
- Tempistiche normative diverse
- Documentazione amministrativa specifica per Paese
- Aggiornamenti delle informazioni sui prodotti
- Obblighi di farmacovigilanza
- Attività di gestione del ciclo di vita
- Aspettative di formattazione dell’invio
Man mano che le aziende si espandono in ulteriori mercati europei e internazionali, mantenere la coerenza tra i registri normativi diventa sempre più importante. Anche piccole differenze tra le presentazioni, le tempistiche o i requisiti amministrativi possono creare ritardi evitabili e ulteriori pressioni normative.
I processi di presentazione ben organizzati aiutano le aziende a mantenere una maggiore visibilità tra le attività normative, riducendo al contempo il rischio di incoerenze tra le autorità.
Per molte aziende farmaceutiche, la gestione di queste responsabilità internamente può diventare ad alta intensità di risorse, in particolare quando le presentazioni coinvolgono più mercati, le aspettative normative in evoluzione e le attività del ciclo di vita in corso.
Navigare nel panorama normativo in evoluzione in Europa
Registrar Corp, esperto di regolamentazione globale, aiuta le aziende farmaceutiche a gestire complesse attività normative europee con maggiore chiarezza e fiducia attraverso i nostri servizi di supporto normativo europeo.
Il nostro team supporta le aziende attraverso:
- Gestione dell’invio in più Paesi
- Supporto per la pubblicazione di eCTD
- Aggiornamenti amministrativi e attività del ciclo di vita
- Organizzazione dei documenti normativi
- Supporto alla gestione di ASMF e CEP
I nostri esperti di regolamentazione aiutano a semplificare la preparazione dell’invio migliorando la coerenza dei documenti, supportando un coordinamento normativo più efficiente e aiutando le aziende a mantenere un maggiore controllo sulla documentazione normativa e sui flussi di lavoro procedurali.
Forniamo anche un accesso sicuro a RegistrarHub, il nostro archivio di documenti online progettato per aiutare le aziende a mantenere l’accesso centralizzato a importanti documenti normativi e documenti di presentazione.
Poiché i quadri normativi europei e internazionali continuano a evolversi, le aziende che investono in processi normativi strutturati e nel supporto esperto alla presentazione sono in una posizione migliore per mantenere la conformità, ridurre la complessità amministrativa e sostenere una crescita sostenibile del mercato in più giurisdizioni.
