
Um ein pharmazeutisches Produkt auf den europäischen Markt zu bringen, ist viel mehr erforderlich als die Vorbereitung eines Zulassungsantrags. Unternehmen müssen das Netzwerk von Agenturen und Organisationen verstehen, die Produktzulassungen, Qualitätsstandards, Lebenszyklusmanagement und Compliance-Verpflichtungen in ganz Europa beeinflussen.
Während die Europäische Arzneimittelagentur (EMA) für viele pharmazeutische Zulassungen weiterhin von zentraler Bedeutung ist, geht der regulatorische Rahmen Europas weit über eine einzige Behörde hinaus. Organisationen wie die Heads of Medicines Agencies (HMA), die European Directorate for the Quality of Medicines & HealthCare (EDQM), Swissmedic und die Medicines and Healthcare Products Regulatory Agency (MHRA) spielen jeweils eine wichtige Rolle bei der Gestaltung der Überprüfung, Überwachung und Wartung von Arzneimitteln während ihres gesamten Lebenszyklus. Zusammen veranschaulichen diese Organisationen, wie sich die europäische Regulierung von unabhängigen nationalen Systemen zu einem koordinierten Ökosystem entwickelt hat, das die regulatorische Aufsicht, die Qualitätsstandards und den Marktzugang in Einklang bringt.
Zusammen veranschaulichen diese Organisationen, wie sich die europäische Pharmaregulierung von einer Sammlung unabhängiger nationaler Systeme zu einem hochgradig koordinierten regulatorischen Ökosystem entwickelt hat, das die Harmonisierung mit der länderspezifischen Aufsicht in Einklang bringt.
Die Entwicklung des europäischen Regulierungsrahmens
Vor den 1990er Jahren wurden pharmazeutische Zulassungen in Europa hauptsächlich von einzelnen nationalen Behörden verwaltet. Jedes Land unterhielt seine eigenen regulatorischen Verfahren, Überprüfungsstandards und Genehmigungszeitpläne, was zu einer erheblichen Komplexität für Unternehmen führte, die Zugang zu mehreren europäischen Märkten suchten.
Als die pharmazeutische Entwicklung zunehmend global wurde, erkannten die Regulierungsbehörden die Notwendigkeit einer stärkeren Zusammenarbeit, stärkerer Qualitätsstandards und effizienterer Überprüfungsprozesse. Dies führte zur Schaffung und Erweiterung mehrerer Organisationen, die die Koordination, Harmonisierung und regulatorische Konsistenz in der gesamten Region unterstützen sollen.
Die Einrichtung der EMA führte zentralisierte Überprüfungswege für bestimmte Medikamente ein. Gleichzeitig verstärkten Kooperationsstrukturen wie die HMA die Zusammenarbeit zwischen nationalen Behörden, während EDQM die Rolle der harmonisierten pharmazeutischen Qualitätsstandards durch das System European Pharmacopoeia and Certificate of Suitability (CEP) erweiterte.
In jüngster Zeit haben regulatorische Entwicklungen wie der Brexit die Landschaft weiter verändert, indem sie eine unabhängige Rolle für die MHRA außerhalb des Rahmenwerks der Europäischen Union geschaffen haben.
Heutzutage müssen Pharmaunternehmen in einem regulatorischen Umfeld navigieren, das mehrere Behörden und Organisationen umfasst, von denen jede unterschiedliche Verantwortlichkeiten und regulatorischen Einfluss hat. 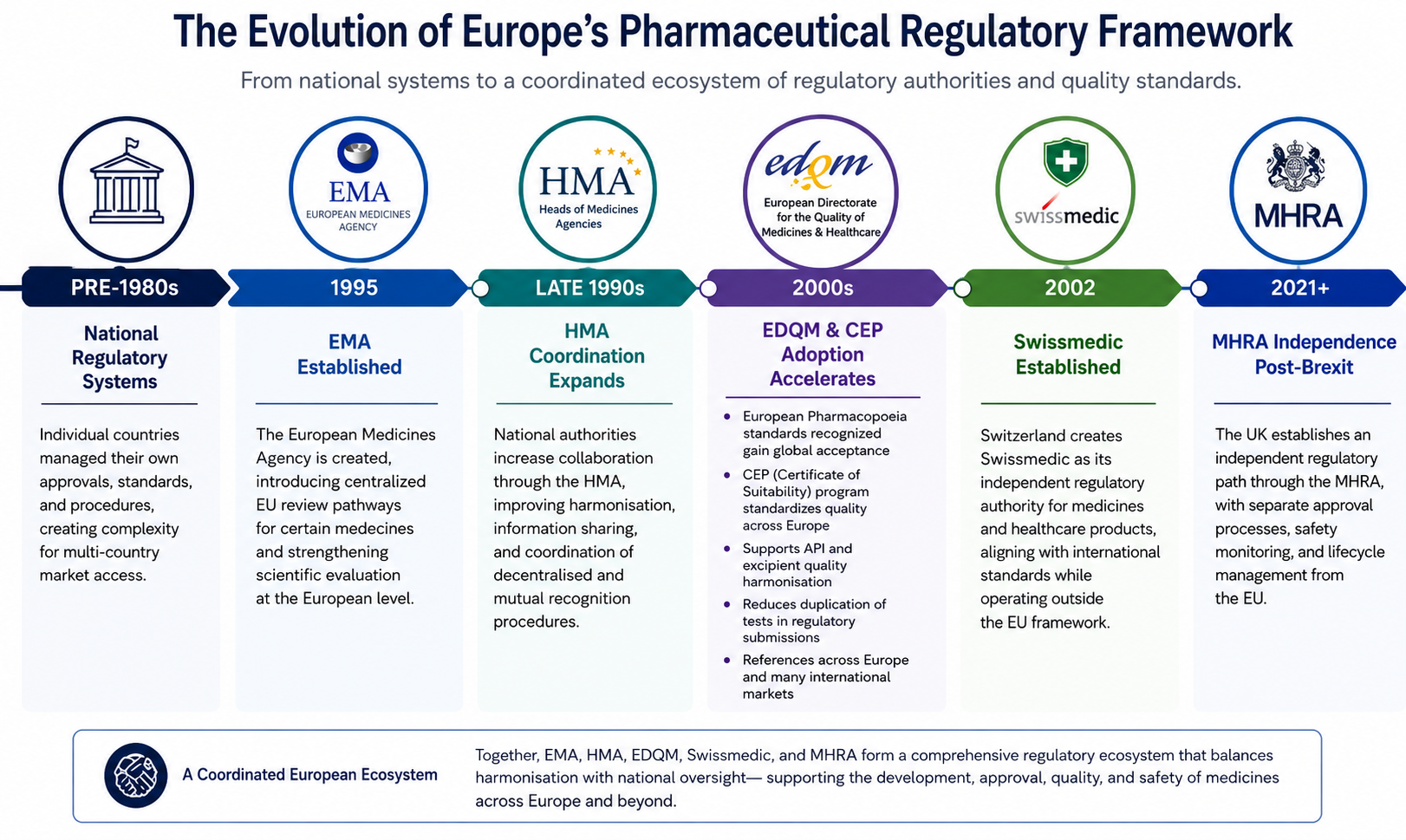
Die Rolle der Heads of Medicines Agencies (HMA)
Die Heads of Medicines Agencies (HMA) sind ein Netzwerk, das sich aus den nationalen zuständigen Behörden zusammensetzt, die für die Regulierung von Arzneimitteln im Europäischen Wirtschaftsraum (EWR) verantwortlich sind.
Im Gegensatz zur EMA ist die HMA keine zentrale Zulassungsbehörde. Stattdessen dient es als kollaborativer Rahmen, der bei der Koordinierung der regulatorischen Aktivitäten zwischen europäischen nationalen Behörden hilft.
Die HMA wurde in den 1990er Jahren formell gegründet, da die europäische regulatorische Zusammenarbeit neben der Einführung dezentralisierter und gegenseitiger Anerkennungsverfahren expandierte. Da immer mehr pharmazeutische Anwendungen mehrere Länder gleichzeitig involvieren, erkannten die Aufsichtsbehörden die Notwendigkeit einer stärkeren Koordination zwischen den nationalen Behörden.
Mit der Zeit ist die HMA zu einem wichtigen Teil der regulatorischen Struktur Europas geworden, indem sie dazu beiträgt, Folgendes zu verbessern:
- Regulatorische Harmonisierung
- Informationsaustausch zwischen Behörden
- Koordination bei dezentralen Verfahren
- Zusammenarbeit bei der Pharmakovigilanz
- Regulatorische Datenstandardisierung
Die Organisation unterstützt auch Arbeitsgruppen und Initiativen, die sich auf die Verbesserung der Konsistenz bei den europäischen regulatorischen Aktivitäten konzentrieren.
Für Pharmaunternehmen spielt die HMA eine wichtige Rolle, um nationalen Behörden dabei zu helfen, sich auf wissenschaftliche Bewertungen, Verfahrensstandards und regulatorische Erwartungen in ganz Europa abzustimmen.
Diese Koordination wird besonders wichtig während:
- Dezentrale Verfahren (DCP)
- Verfahren zur gegenseitigen Anerkennung (MRP)
- Aktivitäten des Lebenszyklusmanagements in mehreren Ländern
- Grenzüberschreitende Koordination der Pharmakovigilanz
Das Verständnis des HMA-Rahmenwerks kann Unternehmen dabei helfen, besser vorauszusehen, wie nationale Behörden in den europäischen Überprüfungsprozessen zusammenarbeiten.
EDQM und CEPs: Die Grundlage der pharmazeutischen Qualitätsstandards
Während sich Behörden wie die EMA, HMA, Swissmedic und die MHRA auf regulatorische Aufsichts- und Marktzulassungsaktivitäten konzentrieren, dient die Europäische Direktion für die Qualität von Arzneimitteln und Gesundheitswesen (EDQM) einer anderen, aber ebenso wichtigen Funktion.
EDQM ist eine unabhängige europäische Institution, die unter dem Council of Europe tätig ist und für die Entwicklung und Pflege des Europäischen Arzneibuchs verantwortlich ist, einer der weltweit am weitesten anerkannten Sammlungen von pharmazeutischen Qualitätsstandards.
Einer der einflussreichsten Beiträge von EDQM zur pharmazeutischen Regulierung ist das Programm Certificate of Suitability (CEP).
Ein CEP zeigt, dass die Qualität einer pharmazeutischen Substanz entsprechend den Anforderungen der entsprechenden Monographie des Europäischen Arzneibuchs ausreichend kontrolliert werden kann. Anstatt wiederholt umfangreiche Substanzdaten an mehrere Zulassungsbehörden zu übermitteln, können Hersteller in ihren Zulassungsanträgen auf ein CEP verweisen.
Im Laufe der Zeit sind CEPs zu einem Eckpfeiler der europäischen pharmazeutischen Regulierung geworden und werden häufig verwendet von:
- Hersteller von Wirkstoffen (API)• Hersteller von Hilfsstoffen (falls zutreffend)• Inhaber der Marktzulassung• Aufsichtsbehörden in ganz Europa• Unternehmen, die internationale Zulassungsanträge verfolgen
Der zunehmende Einsatz von CEPs spiegelt eine wichtige Verschiebung der europäischen Regulierung in Richtung einer stärkeren Harmonisierung der pharmazeutischen Qualitätsstandards wider. Für Unternehmen, die komplexe Lieferketten und Multi-Market-Einreichungen verwalten, können CEPs dazu beitragen, die Konsistenz zu verbessern, Duplikationen zu reduzieren und effizientere regulatorische Überprüfungsprozesse zu unterstützen.
Swissmedic – Unabhängiger regulatorischer Ansatz der Schweiz
Obwohl die Schweiz kein Mitglied der Europäischen Union ist, bleibt Swissmedic eine der angesehensten Arzneimittelzulassungsbehörden Europas.
Swissmedic wurde 2002 nach der Konsolidierung der regionalen Aufsichtssysteme der Schweiz in eine zentrale nationale Behörde gegründet. Seitdem hat die Behörde ihre internationalen Kooperationsbemühungen weiter ausgebaut und gleichzeitig einen unabhängigen regulatorischen Rahmen außerhalb des EU-Systems beibehalten.
Swissmedic ist heute verantwortlich für:
- Prüfung und Zulassung von Arzneimitteln in der Schweiz
- Überwachung der Produktsicherheit
- Durchführung von Inspektionen
- Überwachung der Einhaltung der Herstellungsvorschriften
- Verwaltung von Pharmakovigilanz-Aktivitäten
Die Schweiz ist nach wie vor ein wichtiger Pharmamarkt mit einer starken globalen Branchenpräsenz, was Swissmedic zu einer wichtigen Autorität für Unternehmen macht, die breitere europäische Vermarktungsstrategien verfolgen.
Obwohl Swissmedic mit internationalen Regulierungsbehörden zusammenarbeitet und an Vertrauens- oder Anerkennungswegen teilnehmen kann, bleiben ihre Überprüfungsverfahren unabhängig von der EMA.
Für Pharmaunternehmen bedeutet dies, dass Schweizer Einreichungen häufig eine sorgfältige Planung erfordern:
- Länderspezifische administrative Anforderungen
- Lokale Vertretungspflichten
- Erwartungen an die Produktkennzeichnung
- Schweiz-spezifische Einreichungszeitpläne
- Anforderungen an Sprache und Dokumentation
Unternehmen sollten es vermeiden, davon auszugehen, dass die EU-Genehmigung automatisch die Übereinstimmung mit den regulatorischen Erwartungen der Schweiz garantiert. Die Aufrechterhaltung organisierter Einreichungsstrategien in den Märkten der EU und der Schweiz bleibt für einen effizienten Marktzugang unerlässlich.
Die MHRA – der regulatorische Übergang des Vereinigten Königreichs
Die Medicines and Healthcare Products Regulatory Agency (MHRA) dient als nationale Aufsichtsbehörde für Arzneimittel, Medizinprodukte und Gesundheitsprodukte im Vereinigten Königreich.
Die MHRA spielt seit Langem eine wichtige Rolle bei der europäischen Arzneimittelregulierung. Allerdings haben sich seine Verantwortlichkeiten nach dem Austritt Großbritanniens aus der Europäischen Union deutlich weiterentwickelt.
Vor dem Brexit wurden viele pharmazeutische Zulassungen in Großbritannien innerhalb des breiteren EMA-Rahmens durchgeführt. Seit sie eine unabhängige Aufsichtsbehörde wurde, hat die MHRA separate Genehmigungswege, unabhängige Lebenszyklusmanagementverfahren und neue internationale Zuverlässigkeitsmodelle eingeführt, die die regulatorische Flexibilität auf dem britischen Markt unterstützen sollen.
Heute überwacht die MHRA:
- Marktzulassungen in Großbritannien
- Arzneimittelsicherheit und Pharmakovigilanz
- GMP-Inspektionen
- Beaufsichtigung klinischer Studien
- Überwachung der Einhaltung gesetzlicher Vorschriften
Für Pharmaunternehmen hat der Brexit eine völlig neue regulatorische Dynamik zwischen dem EU- und dem britischen Markt geschaffen.
Organisationen, die jetzt sowohl EU- als auch UK-Kommerzialisierungsstrategien verwalten, müssen Folgendes beibehalten:
- Separate Zulassungsanträge
- Unabhängige Lebenszyklusmanagement-Aktivitäten
- Ausgeprägte Kennzeichnungskonformität
- GB-spezifische regulatorische Fristen
- Parallele Pharmakovigilanz-Koordination
Gleichzeitig entwickelt die MHRA weiterhin internationale Kooperations- und Vertrauenswege, die den Zugang zu Medikamenten beschleunigen und gleichzeitig die Aufsicht über die Aufsichtsbehörden erhalten sollen.
Dieses sich entwickelnde Umfeld hat eine starke regulatorische Koordination für Unternehmen, die in beiden Gerichtsbarkeiten tätig sind, immer wichtiger gemacht.
Warum es wichtig ist, diese Behörden zu verstehen
Obwohl die HMA, Swissmedic und MHRA unterschiedlich arbeiten, spielt jede eine wichtige Rolle im breiteren pharmazeutischen regulatorischen Umfeld Europas.
Zu verstehen, wie sich diese Behörden entwickelt haben und wie sie heute funktionieren, hilft Pharmaunternehmen:
- Aufbau stärkerer Markteintrittsstrategien
- Organisiertere Einreichungen vorbereiten
- Antizipieren Sie länderspezifische Erwartungen
- Verbesserung der Lebenszyklusmanagement-Koordination
- Vermeidbare Einreichungsverzögerungen reduzieren
Da die pharmazeutischen Vorschriften zunehmend über globale Märkte hinweg miteinander verbunden werden, müssen Unternehmen die regionale Harmonisierung mit den länderspezifischen Compliance-Anforderungen in Einklang bringen.
Ohne eine strukturierte regulatorische Strategie können Unterschiede zwischen den Behörden administrative Ineffizienzen, inkonsistente Dokumentation und erhöhte betriebliche Komplexität schaffen.
Häufige Herausforderungen im behördenübergreifenden Regulierungsmanagement
Die Verwaltung von Einreichungen über mehrere europäische Behörden hinweg erfordert häufig, dass Unternehmen Folgendes koordinieren:
- Verschiedene regulatorische Zeitpläne
- Länderspezifische administrative Dokumentation
- Aktualisierung von Produktinformationen
- Pharmakovigilanz-Verpflichtungen
- Lebenszyklus-Management-Aktivitäten
- Erwartungen an die Einreichungsformatierung
Da Unternehmen in zusätzliche europäische und internationale Märkte expandieren, wird die Aufrechterhaltung der Konsistenz über alle regulatorischen Aufzeichnungen hinweg immer wichtiger. Selbst kleine Unterschiede zwischen Einreichungen, Zeitplänen oder administrativen Anforderungen können zu vermeidbaren Verzögerungen und zusätzlichem regulatorischem Druck führen.
Gut organisierte Einreichungsprozesse helfen Unternehmen dabei, eine bessere Transparenz über regulatorische Aktivitäten zu erhalten und gleichzeitig das Risiko von Inkonsistenzen zwischen Behörden zu reduzieren.
Für viele Pharmaunternehmen kann die interne Verwaltung dieser Verantwortlichkeiten ressourcenintensiv werden, insbesondere wenn Einreichungen mehrere Märkte betreffen, sich weiterentwickelnde regulatorische Erwartungen und laufende Lebenszyklusaktivitäten.
Navigation in Europas sich entwickelnder regulatorischer Landschaft
Registrar Corp ist ein erfahrener globaler Experte für regulatorische Angelegenheiten und hilft Pharmaunternehmen, komplexe europäische regulatorische Aktivitäten mit größerer Klarheit und Zuversicht durch unsere europäischen regulatorischen Unterstützungsdienste zu steuern.
Unser Team unterstützt Unternehmen durch:
- Verwaltung von Einreichungen in mehreren Ländern
- eCTD-Publishing-Unterstützung
- Administrative Aktualisierungen und Lebenszyklusaktivitäten
- Organisation regulatorischer Dokumente
- Unterstützung beim ASMF- und CEP-Management
Unsere regulatorischen Experten helfen bei der Vereinfachung der Einreichungsvorbereitung, indem sie die Konsistenz von Dokumenten verbessern, eine effizientere regulatorische Koordination unterstützen und Unternehmen dabei unterstützen, eine stärkere Kontrolle über regulatorische Dokumentationen und Verfahrensabläufe zu behalten.
Wir bieten auch sicheren Zugriff auf RegistrarHub, unser Online-Dokumenten-Repository, das Unternehmen dabei unterstützt, zentralen Zugriff auf wichtige regulatorische Dokumente und Einreichungsunterlagen zu erhalten.
Mit der Weiterentwicklung europäischer und internationaler regulatorischer Rahmenbedingungen sind Unternehmen, die in strukturierte regulatorische Prozesse investieren, und erfahrene Unterstützung bei der Einreichung besser positioniert, um die Compliance aufrechtzuerhalten, die administrative Komplexität zu reduzieren und ein nachhaltiges Marktwachstum in mehreren Gerichtsbarkeiten zu unterstützen.
