
For decades, the regulation of over-the-counter (OTC) drugs in the United States remained largely unchanged, relying on a slow, cumbersome rulemaking process. That shifted fundamentally with the passage of the Coronavirus Aid, Relief, and Economic Security (CARES) Act in 2020, which authorized the Over-the-Counter Monograph Drug User Fee Program (OMUFA).
Today, OMUFA is not just a regulatory hurdle; it is a critical financial and operational requirement for any company manufacturing or distributing OTC monograph drugs for the U.S. market. Failure to understand your facility’s specific classification or missing a narrow fee deadline doesn’t just result in a minor fine—it can lead to your products being legally deemed “misbranded” and your company being placed on the public FDA Arrears List. This public designation effectively halts your ability to do business in the United States, creating an operational paralysis that can take months to resolve.
The Core of OTC Reform: Why OMUFA Exists
Before OMUFA, the FDA’s process for updating OTC monographs was a multi-year “rulemaking” cycle that struggled to keep pace with scientific innovation. OMUFA replaced this with an “administrative order” process, designed to provide the FDA with a sustainable, industry-funded revenue stream to:
- Modernize the OTC Review Process: Moving away from static regulations to a dynamic system that can respond to new safety data in real-time.
- Accelerate Scientific Timelines: Speeding up the review of new active ingredients, dosage forms, and indications that previously languished in administrative backlogs.
- Expand Agency Resources: Funding the hiring of dedicated reviewers, chemists, and inspectors specifically tasked with managing the vast and diverse landscape of OTC monograph products.
In exchange for these fees, the pharmaceutical industry gains a more predictable and efficient regulatory environment. However, this efficiency comes with a strict “pay-to-play” mandate for all facilities involved in the supply chain.
Understanding OMUFA Facility Types and Fees
One of the most common points of confusion for manufacturers is determining which fee applies to their specific business model. Misclassification can lead to significant overpayment or, conversely, underpayment that triggers a compliance audit. The FDA categorizes facilities into two primary types under OMUFA:
1. Monograph Drug Facility (MDF)
An MDF is a foreign or domestic facility that manufactures or processes the “finished dosage form” of an OTC monograph drug. This includes the final physical form of the drug—such as tablets, capsules, or topical creams—intended for consumer use.
- Who Pays: Any facility that physically manufactures, packages, or labels the final product. Even if you are a foreign manufacturer shipping to a U.S. distributor, the MDF fee generally applies to your production site.
- The Cost: MDFs are responsible for the full annual facility fee, which is adjusted yearly to meet the FDA’s statutory revenue targets.
2. OTC Monograph Drug Contract Manufacturing Organization (CMO) Facility
A CMO facility is a facility that manufactures the finished dosage form of an OTC monograph drug but is not the owner of such manufacturing facility nor any affiliate of such owner or facility sells the OTC monograph drug produced at such facility directly to wholesalers, retailers, or consumers in the United States.
- Who Pays: Typically, contract manufacturing organizations fall into this category if they are producing the drug solely on behalf of a brand owner and do not hold the regulatory “order” for the product.
- The Cost: To account for their indirect role in the monograph request process, these facilities generally pay a reduced fee, typically set at two-thirds of the standard MDF fee.
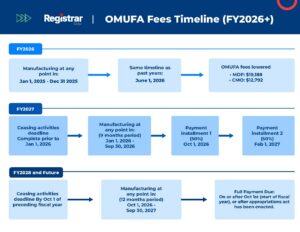
2025–2026 Fee Outlook
The FDA announces OMUFA fees annually, calculated based on their projected operating costs, inflation adjustments, and the number of registered facilities in the system. Because the total revenue target is fixed by law, the individual fee per facility can fluctuate depending on how many companies successfully register and list their products each year.
| Facility Type | FY 2024 Fee | FY 2025 Fee | FY 2026 Fee |
| Monograph Drug Facility (MDF) | $34,166 | $37,556 | $19,188 |
| OTC Monograph Drug CMO Facility | $22,777 | $25,037 | $12,792 |
Note: Fees are mandatory and must be paid in full; the FDA does not offer prorated fees or installments. Final figures are usually published in the Federal Register 45 days before the payment deadline.
The Consequences of Non-Payment: The FDA Arrears List
The most significant and immediate risk of OMUFA non-compliance is the FDA Arrears List. If a facility fails to pay its required user fee within 20 days of the due date, the FDA will move that facility from “good standing” to the public Arrears List.
Deep Implications of Being on the Arrears List:
- Legal Misbranding: Once listed, all OTC monograph drugs manufactured at that facility are legally “misbranded” under Section 502(ff) of the FD&C Act. It is a prohibited act to introduce misbranded drugs into interstate commerce, exposing the company to seizures and injunctions.
- Import Alerts and Detentions: For foreign facilities, being on the Arrears List is a red flag for U.S. Customs and Border Protection (CBP). Shipments are frequently detained at the port of entry, leading to spoiled products, demurrage fees, and broken supply chains.
- Commercial Fallout (Sponsor Risk): Major retailers (such as Walmart, Target, or Amazon) and large-scale distributors monitor the Arrears List as part of their standard vendor compliance. If your facility appears there, your partners may be legally forced to pull your products from their shelves to avoid their own liability for selling misbranded goods.
Navigating OMORs: The Innovation Pathway
Beyond recurring facility fees, OMUFA introduced the Over-the-Counter Monograph Order Request (OMOR). This is the official administrative pathway for companies seeking to innovate within the monograph system, such as adding a new active ingredient, changing a dosage limit, or requesting a new therapeutic indication.
The FDA categorizes these requests into two tiers based on the complexity of the scientific review required:
- Tier 1 OMOR: Reserved for complex, resource-intensive requests. This includes adding a new active ingredient to a monograph, creating a brand-new therapeutic category, or changing the “standard” indication for an entire class of drugs. These fees are substantial, currently exceeding $500,000 per request, to cover the exhaustive clinical data review required.
- Tier 2 OMOR: Covers more focused, modest changes. This might include reordering the safety warnings on a label, adding a minor dosage form that doesn’t require new clinical safety studies, or clarifying specific use instructions. These fees currently exceed $100,000 per request.
Key Deadlines and Compliance Actions
OMUFA compliance is an annual lifecycle that requires proactive monitoring. Missing a single window can trigger the Arrears List process. To maintain good standing, your team must hit these specific milestones:
- Annual Registration & Listing Update: You must verify and update your facility registration and drug listings in the FDA’s official submission portals. If your registration is inaccurate or fails to correctly identify your products as monograph drugs, your fee invoice will be incorrect.
- Fee Announcement Monitoring: The FDA typically publishes the upcoming fiscal year’s fees in the Federal Register. This is the moment to secure your budget for the upcoming payment.
- The 45-Day Payment Window: Once the fees are announced, you have a very narrow window—typically 45 days—to ensure the funds are cleared by the FDA’s bank. Keeping a close watch on these shifting deadlines is essential to avoid being placed on the Arrears List.
See this detailed OMUFA Flowchart for more information.

How Registrar Corp Secures Your OMUFA Compliance
Managing FDA user fees involves more than just processing a payment. It requires a nuanced understanding of facility classifications and a precise determination of whether your products fall under the “monograph” system or require a different pathway like an NDA or ANDA.
Registrar Corp provides a comprehensive regulatory shield to ensure your OTC business remains uninterrupted:
- Facility Classification Audit: We conduct a deep-dive analysis of your manufacturing operations to ensure you are classified correctly. We have helped numerous facilities avoid overpaying by correctly identifying them as CMOs rather than MDFs.
- Secure Payment Facilitation: We manage the complex electronic transfer of fees to the FDA’s specific accounts, providing you with verified proof of payment that can be shared with sponsors and customs brokers to confirm your status.
- Registration & Listing Maintenance: We manage your annual facility registration and drug listing updates. If the FDA’s internal data regarding your monograph products doesn’t perfectly match your facility registration, it can lead to administrative errors that jeopardize your standing on the Arrears List—we prevent these issues before they happen.
- Crisis Intervention (Arrears Resolution): If your facility is already on the Arrears List or is facing an import detention, we provide immediate intervention to settle back fees, correct registration errors, and petition the FDA for removal from the list to restore your market access.
Protect your U.S. market access and maintain the confidence of your commercial partners. Contact Registrar Corp today for expert, end-to-end OMUFA assistance.






